百科
文章
話題
鳥氨酸缺乏症
鳥氨酸缺乏症全稱為鳥氨酸氨甲醯轉移酶缺乏致高氨血癥,是一種X連鎖不完全顯性遺傳病。此病患者可在任何年齡發病,其主要表現為高氨血癥的一系列症狀,如新生患兒會出現餵養困難、呼吸急促和昏睡等,成人患者多表現為慢性神經系統損傷,而有生育需求的患者建議通過第三代試管技術生出健康寶寶。

本病全稱為鳥氨酸氨甲醯轉移酶缺乏致高氨血癥(ornithine transcarbamylase deficiency, hyperammonemia due to; OMIM 311250),是因鳥氨酸氨甲醯轉移酶活性降低或缺乏導致的高氨血癥。鳥氨酸氨甲醯轉移酶缺乏症是尿素迴圈障礙中最常見的型別,美國患病率約為5.9/10萬,日本約為1.3/10萬,芬蘭約為1.6/10萬,義大利約為1.4/10萬。我國的患病率不詳。
臨床表現
患者可於任何年齡發病,主要表現為高氨血癥的一系列症狀,主要分為兩型:新生兒期急性起病型和新生兒後起病型,後者亦即遲髮型。
大部分男性患者常於新生兒期發病,出生時可無異常,數天內開始出現易激惹、餵養困難、呼吸急促和昏睡等表現,並迅速發展為痙攣、昏迷和呼吸衰竭,若不及時治療,常在新生兒期死亡,倖存者多遺留嚴重的智力損害。部分患兒表現為遲髮型,多於嬰幼兒期起病,症狀相對較輕,臨床表現更為多變,如肝大、反覆發作的癲癇、生長髮育障礙及行為異常等。
兒童和成人期發病者常表現為慢性神經系統損傷,以各種行為異常、精神錯亂、煩躁易怒和發作性嘔吐為特徵。
雜合子女性攜帶者多數終身無症狀,少數有發病,其發病年齡及臨床表現的個體差異性明顯,即可表現為嚴重的新生兒期急性起病型,又可表現為成年期發病型。

相關基因介紹
鳥氨酸氨甲醯轉移酶缺乏症,屬X連鎖不完全顯性遺傳,編碼鳥氨酸氨甲醯轉移酶(ornihtine transcarbamylase, OTC)的基因OTC位於Xp21.1,全長75 968bp,有10個外顯子,編碼的OTC含354個氨基酸。迄今已報道了340多種突變型,其中,80%為點突變,其他包括插入和缺失等。
因OTC基因發生突變,鳥氨酸氨甲醯轉移酶活性喪失或低下,氨甲醯與鳥氨酸合成瓜氨酸受阻,導致血氨甲醯磷及血氨增高、瓜氨酸降低。氨甲醯磷酸增高導致其旁路代謝產物乳清酸及尿嘧啶增多。血氨增高干擾腦細胞的能量代謝,可引起腦內興奮性神經遞質減少,抑制性神經遞質增多,引起腦損傷。
案例分享
基本資訊
家系特點:父親正常,母親為攜帶者,攜帶OTC基因突變(c.917G>C),先證者男性為患者,尋求PGD助孕。
檢測父母雙親、先證者1個,胚胎12個(JQ1、JQ2、JQ3、JQ4、JQ5、JQ6、JQ7、JQ8、JQ9、JQ10、JQ11、JQ12)。
檢測方法
三代試管嬰兒S-PGD——三重防護
JBRH解決方案檢測特點
- 檢出率高,適用範圍廣;
- 檢測結果準確,能夠有效檢測到重組,可無先證者;
- 靶向捕獲設計,方案靈活,便於臨床成本控制。
S-PGD檢測結果
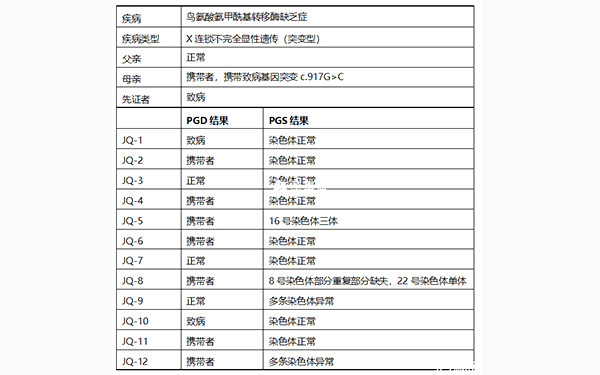
經PGD檢測發現:12個胚胎中,2個胚胎(JQ-1、JQ-10)為致病胚胎,7個胚胎(JQ-2、JQ-4、JQ-5、JQ-6、JQ-8、JQ-11、JQ-12)為攜帶者胚胎,3個胚胎(JQ-3、JQ-7、JQ-9)為正常胚胎。
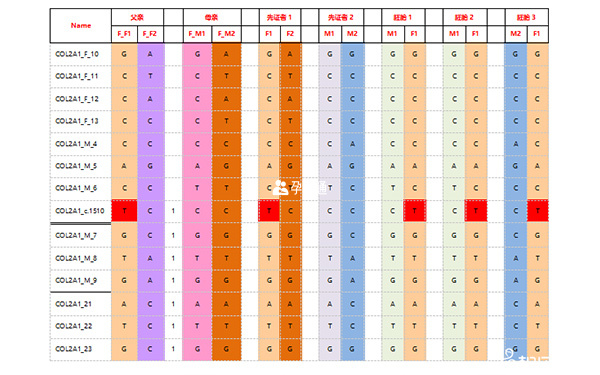
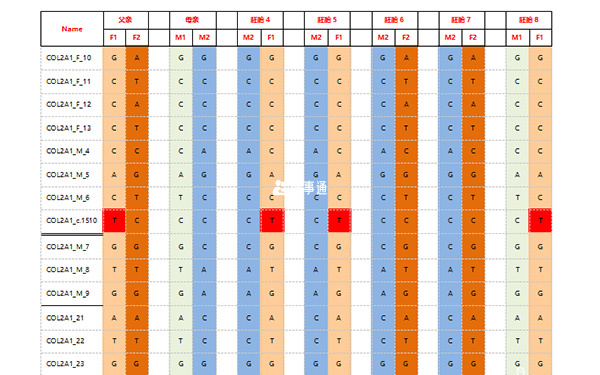
經PGS檢測發現:12個胚胎中,胚胎JQ-1、JQ-2、JQ-3、JQ-4、JQ-6、JQ-7、JQ-10、JQ-11的染色體正常;胚胎JQ-5為16號染色體三體;胚胎JQ-8為8號染色體部分重複部分缺失,22號染色體單體;胚胎JQ-9和JQ-12都多條染色體異常。
JBRH基於SNP連鎖分析的S-PGD解決方案優勢
做三代試管嬰兒臨床意義
▪更適用於臨床使用:對每種疾病精細化設計,檢測致病位點,可檢測到重組,檢測準確度更高;
▪分析結果更靈敏:測序不但能夠利用設計好的SNP,還能發現新的SNP並加以利用,提高了重組斷點的檢測靈敏度;
▪幫助減少先天性無虹膜症患兒的出生,有利於優生優育。
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜