百科
文章
話題
苯丙酮尿症
苯丙酮尿症(PKU)是一種常染色體隱性遺傳疾病,其臨床表現不均一,主要臨床特徵為智力低下、精神神經症狀、溼疹、面板抓痕徵及色素脫失和尿液中鼠氣味等。由於有該病有一定的遺傳率,因此建議通過第三代試管嬰兒技術助孕生育健康的孩子。
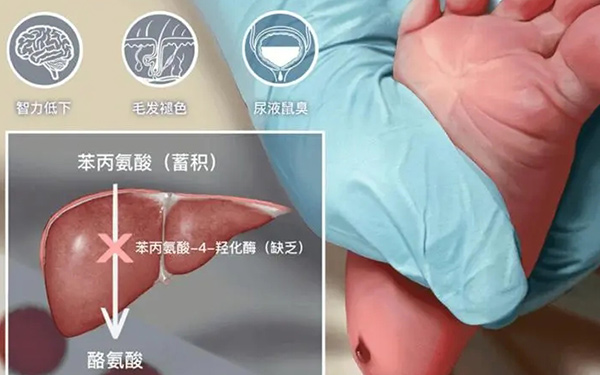
苯丙酮尿症(phenylketonuria, PKU)是由於肝臟苯丙氨酸羥化酶(phenylalaninehydroxylase, PAH)缺乏或活性減低而導致苯丙氨酸代謝障礙的一種遺傳性疾病。在遺傳性氨基酸代謝缺陷疾病中最常見的型別,遺傳方式為常染色體隱性遺傳,中國人群發病率約為1/14000。

臨床表現不均一,主要臨床特徵為智力低下、精神神經症狀、溼疹、面板抓痕徵及色素脫失和尿液中鼠氣味等,腦電圖異常。如果能得到早期診斷和早期治療,則前述臨床表現可不發生,智力正常,腦電圖異常也可得到恢復。
苯丙酮尿症的病因主要為:苯丙氨酸羥化酶和輔因子的缺失,導致苯丙氨酸代謝障礙,產生苯丙酮尿症。
苯丙酮尿症相關基因介紹
PKU為常染色體隱性遺傳,致病基因為PAH基因,位於12號染色體長臂(12q23.2),基因長度86277bp。PAH基因是割裂基因,編碼區包含13個外顯子,被不編碼的12個內含子所分隔。轉錄時剪接內含子形成僅包含外顯子的1353bp的單鏈,即包含451個密碼子的可譯框。mRNA翻譯成含451個氨基酸殘基的酶單體,單體聚合成有功能的PAH酶。
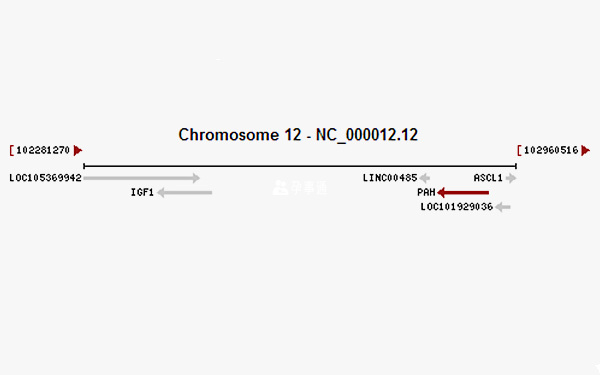
PAH通常以具有催化活性的四聚體形式存在,四聚體是由兩個二聚體組成,PAH依據pH值不同維持二聚體和四聚體的平衡。PAH酶單體由3部分組成:1)N末端調節區(1~142殘基);2)催化區(143~410殘基);3)C末端的四聚體區(411~452殘基)。
PAH基因的微小突變即可引起PKU。最常見的PAH突變中的某些突變即發生於催化區和四聚體區的交界處,即外顯子或內外顯子的剪接區,嚴重影響PAH酶基因的轉錄和翻譯,導致蛋白質的異常摺疊、聚合,以及加速酶的降解,從而影響PAH酶催化活性。迄今已經發現700多種PAH基因突變,中國人群中已報道150多種以上基因突變。
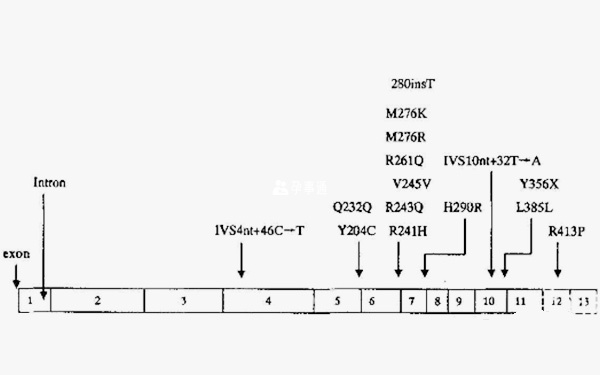
PAH突變特點:
- 1. 突變位置多變:所有外顯子、內含子、5’-UTR和3’-UTR區均發現突變;
- 2. 變型別多樣:有錯義突變、小缺失、剪接位點突變、沉默突變、無義突變、小插入、大片段插入罕見;
- 3. 突變呈明顯異質性:不同種族和地區人群之間PAH酶基因座突變部位及分佈具有較大差異。
苯丙酮尿症三代試管案例分享
基本資訊
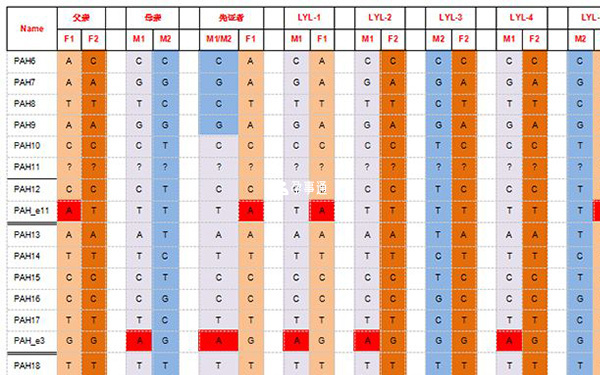
家系特點:該家系先證者發生重組,根據資料情況分析:推測先證者發生重組是由於該基因來自母親的兩條鏈發生重組;
其父親攜帶c.1197 A>T突變位點,母親攜帶c.331 C>T突變位點,申請PGD助孕;
檢測父母雙親、先證者、胚胎5個(LYL-1、LYL-2、LYL-3、LYL-4、LYL-5)。
苯丙酮尿症的檢測方法
三代試管嬰兒S-PGD——三重防護
JBRH解決方案檢測特點
- 檢出率高,適用範圍廣;
- 檢測結果準確,能夠有效檢測到重組,可無先證者;
- 靶向捕獲設計,方案靈活,便於臨床成本控制。
苯丙酮尿症S-PGD檢測結果

經檢測發現:5個胚胎中,1個胚胎(LYL-3)為正常胚胎,4個胚胎(LYL-1、LYL-2、LYL-4、LYL-5)為致病胚胎。結合PGS結果,建議移植LYL-2號胚胎。
JBRH基於SNP連鎖分析的S-PGD解決方案優勢
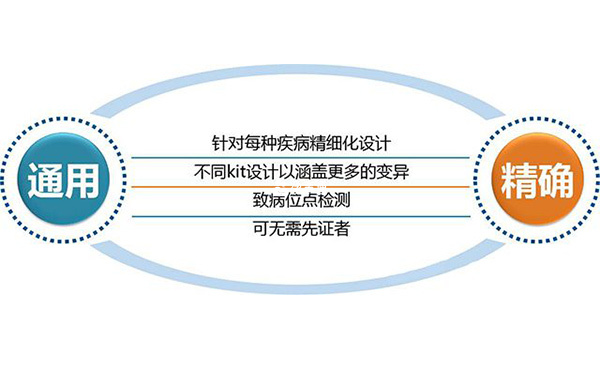
苯丙酮尿症做三代試管的臨床意義:
▪更適用於臨床使用:對每種疾病精細化設計,檢測致病位點,可檢測到重組,檢測準確度更高;
▪分析結果更靈敏:測序不但能夠利用設計好的SNP,還能發現新的SNP並加以利用,提高了重組斷點的檢測靈敏度;
▪幫助減少先天性無虹膜症患兒的出生,有利於優生優育。
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜