百科
文章
話題
神經纖維瘤病
神經纖維瘤病是(NF)一種常染色體顯性遺傳病,根據臨床表現、細胞生物學和分子生物學特點可將其分為Ⅰ型和Ⅱ型,其區別在於是否出現雙側聽神經瘤。雖然神經纖維瘤多數屬於良性腫瘤,只有少部分是惡性的情況,但是為了徹底阻斷此病遺傳給下一代,建議通過第三代試管嬰兒技術助孕。
神經纖維瘤病(neurofibromatosis, NF),常染色體顯性遺傳病,源於神經嵴細胞異常導致的多系統損害疾病。年發病率約為1/20000。根據臨床表現,細胞生物學和分子生物學特點將其分為Ⅰ型和Ⅱ型。
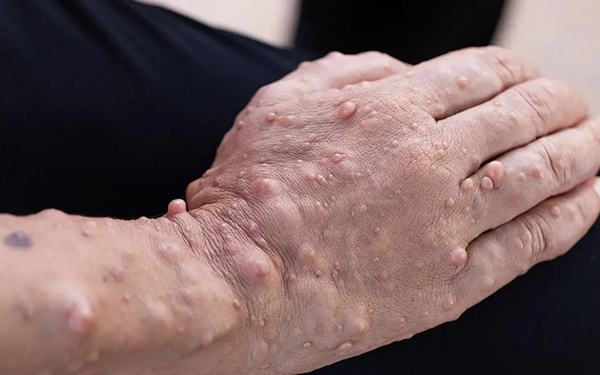
神經纖維瘤病Ⅰ型(NF1,OMIM 162200)
NF1稱為周圍神經纖維瘤病(俗稱“橡皮人”)。
臨床特徵:多發的牛奶咖啡斑(>6個,青春期前患者最大徑>0.5cm)、腋下和腹股溝雀斑、散在的多發面板神經纖維瘤(>3個)和虹膜Lisch結節(錯構瘤),有半數以上的患者會表現為學習困難。比較少見但更為嚴重的症狀還包括網狀神經纖維瘤、視神經和其他中樞神經系統的神經膠質瘤、惡性外周神經鞘腫瘤、脊柱側凸,脛骨發育異常和血管病。50%-70%有家族遺傳史,發病率為1/2500-1/3000新生兒。NF1突變率非常高,突變基因頻率為13-25/10萬配子。本病基因突變率為4.3×10-5。國內統計母系遺傳是父系的2.1倍。NF1患者有1/2的風險將突變傳遞給子女。
防治:廣泛的面板及皮下腫瘤無需特殊治療。如顱內或椎管內的單發腫瘤以及周圍神經腫瘤迅速長大或壓迫神經者,應手術切除,以解除壓迫或防止惡變。
神經纖維瘤病Ⅱ型(NF2,OMIM 101000)
NF2,即雙側聽神經瘤病,有時也被稱為“中央神經纖維瘤病”。
臨床特徵:雙側聽神經瘤、腦膜瘤和脊髓背根神經鞘瘤,但面板損傷或神經纖維瘤少見。慢性起病,逐漸進展,先出現眩暈,耳鳴,耳聾等前庭及耳蝸神經的症狀,後枕部疼痛不適,鄰近腦神經受損表現為面部疼痛,感覺減退,面肌抽搐,周圍性面癱。小腦共濟失調。顱高壓症狀。吞嚥困難和飲水嗆咳。部分患者可伴有面板,皮下組織,周圍神經和脊髓的多發性神經纖維病,面板牛奶咖啡斑和先天性骨骼畸形。NF2發病率為1/20000新生兒。
防治:NF2手術治療效果差,不宜手術。小的腫瘤可採用聚焦放療,大的腫瘤威脅到生命,可考慮手術治療。
相關基因介紹
具有遺傳異質性的NF主要有2個致病基因NF1和NF2,分別導致NF1和NF2。
NF1是常染色體顯性遺傳病,完全外顯,半數患者是基因新生突變(突變率為1/10000),沒有家族史。致病基因為位於17q11.2的NF1基因,為腫瘤抑制基因,編碼神經纖維蛋白。NF1基因長289701bp,由60個外顯子組成。最長的一個轉錄本(NM_000267)包含58個外顯子,編碼2839個氨基酸。已有的研究表明,NF1基因突變形式多樣,有染色體異常,全基因缺失、多個外顯子缺失,插入突變、無義突變、錯義突變和內含子突變。但突變多為微小改變(點突變或微小缺失/重複),4%-5%的患者為NF1全基因缺失。多數散發性突變來自父系。小的突變隨機分佈在整個NF1基因上,這些突變多導致蛋白截短。
NF2屬常染色體顯性遺傳,約一半以上患者為新突變者。致病基因為位於22q11.2上編碼膜突樣蛋白的NF2基因,為腫瘤抑制基因基因。NF2基因含17個外顯子,轉錄產物為神經膜細胞素,被認為是肌動蛋白相關蛋白,在細胞骨架和細胞膜之間起連線作用,該蛋白的缺陷和失活使細胞生長失控。
案例分享
基本資訊
家系特點:父親為NF1患者,母親正常,無先證者,尋求PGD助孕。
檢測父母雙親、胚胎12個(PLM1、PLM2、PLM3、PLM4、PLM5、PLM6、PLM7、PLM8、PLM9、PLM10、PLM11、PLM12)。
檢測方法:以基因NF1基因為目標區域,選擇高度緊密連鎖的SNP位點作為遺傳標記,在基因上下游擴大範圍分方案進行單體型構建,合併方案的位點進行單體型分析。
神經纖維瘤病的檢測方法
三代試管嬰兒S-PGD——三重防護
JBRH解決方案檢測特點
- 檢出率高,適用範圍廣;
- 檢測結果準確,能夠有效檢測到重組,可無先證者;
- 靶向捕獲設計,方案靈活,便於臨床成本控制。
S-PGD檢測結果
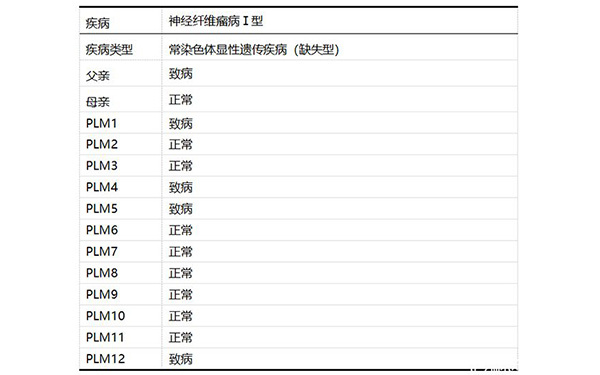
經檢測發現:12個胚胎中,8個胚胎(PLM2、PLM3、PLM6、PLM7、PLM8、PLM9、PLM10、PLM11)為正常胚胎,4個胚胎(PLM1、PLM4、PLM5、PLM12)為致病胚胎。
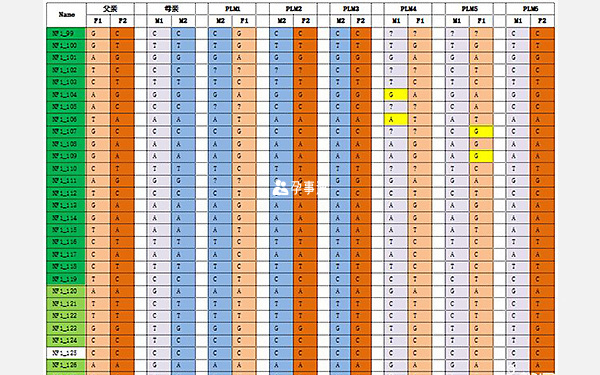
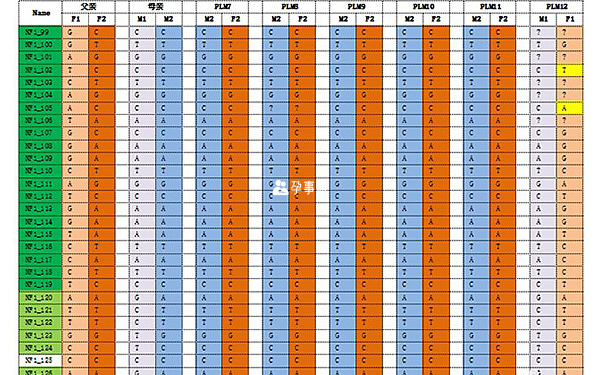
進行PGS發現:胚胎PLM2的16號染色體長臂存在重複;胚胎PLM8的16號染色體單體;胚胎PLM10的4號染色體單體;胚胎PLM11的14號染色體異常。其餘胚胎未發現染色體拷貝數增加/缺失。PGD+PGS綜合分析:胚胎PLM6、PLM7、PLM9既無染色體拷貝數異常,也無NF1致病基因。
JBRH基於SNP連鎖分析的S-PGD解決方案優勢
神經纖維瘤病做三代試管嬰兒的臨床意義:
▪更適用於臨床使用:對每種疾病精細化設計,檢測致病位點,可檢測到重組,檢測準確度更高;
▪分析結果更靈敏:測序不但能夠利用設計好的SNP,還能發現新的SNP並加以利用,提高了重組斷點的檢測靈敏度;
▪幫助減少先天性無虹膜症患兒的出生,有利於優生優育。
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜