百科
文章
話題
脊髓性肌萎縮症
脊髓性肌萎縮症(SMA)是一種由控制肌肉運動的運動神經元缺失引起的遺傳性疾病,其特徵是用於運動的肌肉(骨骼肌)虛弱和萎縮,且肌肉萎縮會隨著年齡的增長而加劇。由於SMA是隱性的遺傳病,所以SMA攜帶夫妻,最好通過三代試管嬰兒技術篩查健康的胚胎,從而阻斷該病的遺傳。

脊髓性肌萎縮症又稱脊肌萎縮症(SMA),它是一種常見的罕見病,臨床表現的症狀為肌無力、肌萎縮,是一類由脊髓前角運動神經元變性導致肌無力、肌萎縮的常染色體隱性遺傳病。

脊髓性肌萎縮症
脊髓性肌萎縮是一種遺傳性疾病,其特徵是用於運動的肌肉(骨骼肌)虛弱和萎縮,是由控制肌肉運動的運動神經元這種特殊神經細胞的缺失引起的,肌肉萎縮通常隨著年齡的增長而加劇。
有多種不同型別的脊髓性肌萎縮是由相同基因的改變引起的,其常見症狀如進行性肌痙攣等。
1、0型SMA: 在出生時就很明顯,是最嚴重的一種SMA。致病的嬰兒在子宮時就會出現胎動過少的現象,從而導致這些嬰兒出生時往往伴隨關節畸形、肌張力弱、呼吸衰竭(呼吸肌萎縮)甚至先天性心臟缺陷。這導致他們往往無法活過嬰兒期。
2、I型SMA: 也稱為威德森-霍夫曼綜合徵(werdnig- hoffmann syndrome),這是SMA的常見型別。這是一種嚴重的肌肉無力症,在出生時或出生後的頭幾個月就很明顯,致病兒童無法控制他們的頭部運動,也無法獨自坐著。這種型別的兒童通常會有吞嚥問題,導致餵養困難,從而出現發育不良。他們還會因為呼吸肌萎縮導致鐘形胸和呼吸困難。大多數I型SMA的兒童由於呼吸衰竭而死亡。
3、II型SMA: 也稱為杜博維茨病(Dubowitz disease),其特徵是6-12個月時的肌無力,起初這種型別的兒童可以在沒有支撐的情況下坐著,然而,隨著肌肉無力不斷惡化,這些兒童必須得到支撐才能坐著,如果沒有支撐,甚至不能站立或行走。他們的手指經常會不由自主地顫抖,脊柱彎曲(脊柱側彎),呼吸肌無力導致呼吸衰竭最終可能危及生命,II型SMA患者的壽命在20-30歲之間。
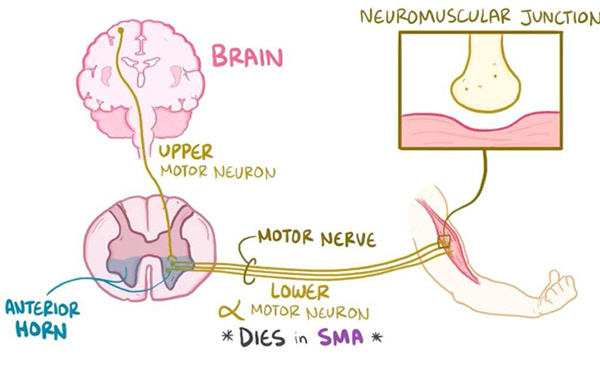
4、III型SMA: 也稱為庫格伯格-維蘭德病( Kugelberg-Welander disease)通常在兒童早期就會引起肌肉無力,患有這種疾病的兒童可以獨立站立和行走,但隨著時間的推移,行走和站立會變得越來越困難,最終只能依靠輪椅。III型SMA患者由於沒有呼吸肌無力,所以壽命與常人無異。
5、IV型SMA: 這類患者非常罕見,其肌無力現象通常在成年時才發生,患者通常會出現輕微到中度的肌無力和輕微的呼吸問題,一般認為,IV型SMA患者的預期壽命和正常人一樣。
全世界每8,000到10,000人中就有1人患有SMA。I型SMA是最常見的型別,約佔所有病例的一半,其次是II型和III型,而0型和IV型非常少見。
致病原因
SMN1基因突變導致上述所有型別SMA的發生,而SMN2基因的拷貝數變化,則決定了SMA的嚴重程度,所以一般通過檢測SMN1和SMN2,就可以提前預知SMA的疾病型別。
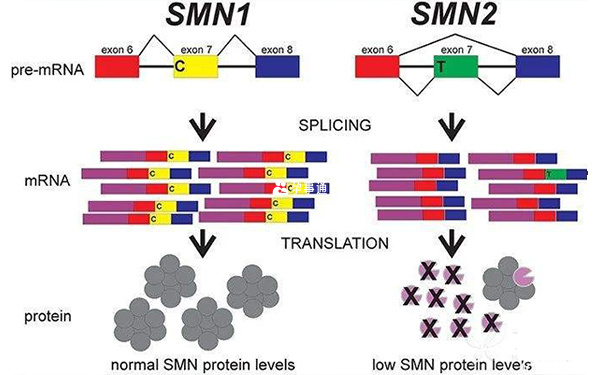
SMN1和SMN2基因都提供製造一種叫做存活運動神經元(SMN)蛋白的指令。正常情況下,大多數功能性SMN蛋白由SMN1基因產生,少量由SMN2基因產生。SMN蛋白是一種被稱為SMN複合體的蛋白質,它對運動神經元的維持非常重要。運動神經元傳遞來自大腦和脊髓的訊號,告訴骨骼肌緊張(收縮),從而使身體運動。
患有SMA的患者都有SMN1基因缺失,而這會影響SMN蛋白的產生。缺乏SMN蛋白會導致訊號無法在大腦和肌肉之間傳遞,最終造成運動神經元死亡。肌肉的收縮離不開大腦的訊號,許多骨骼肌由於接受不到訊號,只會逐步變弱、萎縮。
遺傳規律
SMA是一種常染色體隱性遺傳,這意味著患者的父母都攜帶有突變的SMN1基因。父母通常不會變現出相關疾病症狀,而他們的後代有25%的機率致病。
治療方法
FDA已經批准了兩種治療SMA的藥物:nusinersen (Spinraza)和onasemnogene abeparvoveci -xioi (Zolgensma)。這兩種藥物都是典型的基因治療藥物,是通過調節SMN1和SMN2基因,使其產生更多的SMN蛋白,讓身體能夠提供傳遞大腦訊號的指令,從而幫助患者控制肌肉運動。
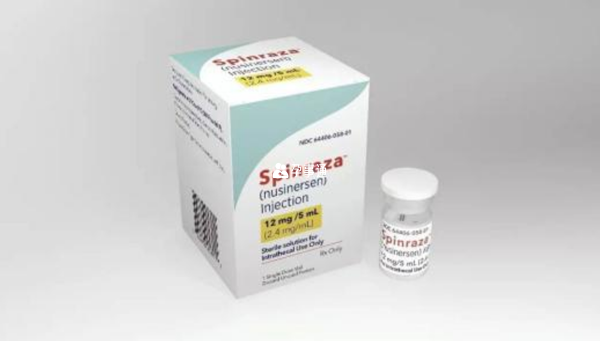
其中Nusinersen主要調節SMN2基因,適合全年齡的SMA患者使用,而Onasemnogene abeparvovec-xioi主要調節SMN1基因,適合2歲以下兒童使用。
生育干預
由於SMA是隱性的遺傳病,有些人表面上是正常的,其實是該基因病的攜帶者,那麼該如何知道是否攜帶呢?目前只能通過基因檢測來完成,如果無法確認自己是否攜帶有SMN1和SMN2基因異常。
如果父母是SMA的攜帶者,又該如何阻斷SMA的遺傳?由於SMA是隱性的遺傳病,可以通過三代試管嬰兒技術篩查健康的胚胎,從而阻斷該病的遺傳,目前國際先進分子診斷技術,可以阻斷已知所有的單基因疾病包括SMA。
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜