百科
文章
話題
7號染色體異常
染色體7跨越約1.59億個DNA構建塊(鹼基對),可能包含900到1,000個基因,佔細胞總DNA的5%以上。7號染色體一旦出現異常情,將會出現FOXP2相關的言語和語言障礙、Greig cephalopolyyndylyy綜合症、羅素銀綜合徵、Saethre-Chotzen綜合徵以及Williams綜合症等疾病。

導航
人類通常在每個細胞中有46條染色體,分為23對。7號染色體的兩個拷貝,一個從每個親本遺傳的拷貝,形成一對。染色體7跨越約1.59億個DNA構建塊(鹼基對),佔細胞總DNA的5%以上。
識別每條染色體上的基因是遺傳研究的一個活躍領域。由於研究人員使用不同的方法來預測每條染色體上的基因數量,因此估計的基因數量會有所不同。染色體7可能包含900到1,000個基因,這些基因提供製造蛋白質的說明。這些蛋白質在體內發揮著各種不同的作用。
7號染色體圖
遺傳學家使用稱為idiograms的圖表作為染色體的標準表示。影象顯示染色體的相對大小及其條帶圖案,這是當染色體用化學溶液染色然後在顯微鏡下觀察時出現的暗帶和亮帶的特徵圖案。這些條帶用於描述每條染色體上基因的位置。
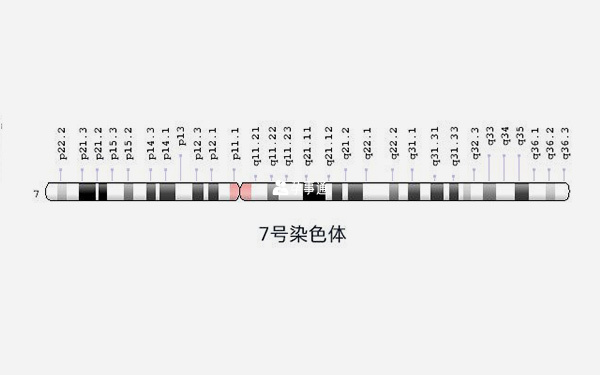
7號染色體異常會出現四種相關病症
FOXP2相關的言語和語言障礙
FOXP2相關的言語和語言障礙影響染色體7的幾種不同變化可導致與FOXP2相關的言語和語言障礙。這些變化涉及含有FOXP2基因的7號染色體長(q)臂區域。與FOXP2相關的言語和語言障礙是一種罕見的病症,它影響從兒童早期開始的言語和語言的發展。在一些受影響的個體中,語言和語言問題是這種情況的唯一特徵。其他人也延遲了運動技能的發展,例如步行和繫鞋帶,以及自閉症譜系障礙,這些疾病的特點是溝通和社互動動受損。
作為FOXP2相關言語和語言障礙的所有遺傳變化破壞了FOXP2的活性,FOXP2是正常言語和語言發展的關鍵基因。一些與FOXP2相關的言語和語言障礙的個體具有去除7號染色體的一小部分的缺失,包括FOXP2基因和幾個相鄰基因。患有這種病症的其他人在FOXP2基因內部具有突變。不常見的是,FOXP2從7號染色體的結構的重排-相關言語和語言障礙的結果(例如,易位),或從來自每一個親從母親而不是一個繼承7號染色體的兩個拷貝(一種稱為母體單親二體或母體UPD現象,下面用Russell-Silver綜合徵更詳細地描述。目前尚不清楚如何使染色體7的兩個母本拷貝影響FOXP2基因的活性。

有時與FOXP2相關的言語和語言障礙相關的其他特徵,包括延遲的運動發育和自閉症譜系障礙,可能是由於染色體7上其他基因的變化引起的。例如,在染色體7缺失的受影響個體中,損失的FOXP2被認為擾亂言語和語言發育,而附近的基因的損失佔其他症狀和體徵。7號染色體母體UPD患者的FOXP2相關言語和語言障礙是Russell-Silver綜合徵的一個較大病症的一部分。
Greig cephalopolyyndylyy綜合症
這是一種影響肢體,頭部和麵部發育的疾病。這些染色體變化涉及包含GLI3基因的染色體7的短(p)臂區域。該基因在出生前在許多組織和器官的發育中起重要作用。
在某些情況下,Greig cephalopolysyndactyly綜合症是由染色體7和另一條染色體之間遺傳物質的重排(易位)引起的。其他病例是由染色體7的短臂中幾個基因(包括GLI3)的缺失引起的。多個基因的丟失可導致這種疾病的更嚴重形式,稱為Greig cephalopolyyndylyyly連續基因缺失綜合徵。患有這種形式的疾病的人具有涉及四肢,頭部和麵部的特徵性發育問題,以及癲癇發作,發育遲緩和智力殘疾。
羅素銀綜合徵
這是一種罕見病症,其特徵是生長緩慢,面部特徵明顯,發育遲緩,語言和語言問題以及學習障礙。
人們通常從母親那裡繼承每個染色體的一個副本,從父親那裡繼承一個副本。對於大多數基因,兩個拷貝都在細胞中表達或“開啟”。然而,對於某些基因,只表達了從一個人的父親(父親副本)遺傳的拷貝。對於其他基因,僅表達從人的母親遺傳的拷貝(母本)。基因表達的這些親本特異性差異是由稱為基因組印記的現象引起的。染色體7含有一組通常經歷基因組印記的基因; 這些基因中的一些僅在母體拷貝上有活性,而其他基因僅在父本拷貝上有活性。

在7%至10%的Russell-Silver綜合徵病例中,人們從母親(母親UPD)繼承了7號染色體的拷貝而不是每個父母的一份拷貝。母體UPD導致人們有兩個印跡基因的活躍拷貝,沒有其他的活躍拷貝。在這些病例中,7號染色體上活躍的母體和父本基因的不平衡是這種疾病的體徵和症狀的基礎。
Saethre-Chotzen綜合徵
這種罕見病症的特徵是某些顱骨過早融合(顱縫早閉),這可防止顱骨正常生長並影響頭部和麵部的形狀。染色體變化涉及包含TWIST1基因的7號染色體短(p)臂區域。該基因在頭部,面部和四肢的早期發育中起重要作用。
導致Saethre-Chotzen綜合徵的染色體異常包括7號染色體與另一條染色體之間遺傳物質的易位,7號染色體內遺傳物質的重排(反轉),或稱為環狀染色體7的異常環狀結構的形成。環染色體當染色體在兩個地方破裂並且染色體臂的末端融合在一起形成圓形結構時發生。這些染色體變化中的每一個都改變或刪除TWIST1基因,並且還可能影響附近的基因。

當Saethre-Chotzen綜合徵由染色體缺失而不是TWIST1基因內的突變引起時,受影響的兒童更有可能患有智力殘疾,發育遲緩和學習困難。這些特徵通常在Saethre-Chotzen綜合徵的典型病例中沒有出現。研究人員認為,7號染色體短臂上的其他基因的丟失可能是造成這些額外特徵的原因。
Williams綜合症
是由Williams-Beuren臨界區遺傳物質的缺失引起的(如上所述)。研究人員認為,威廉姆斯綜合徵的特徵,包括輕度至中度智力殘疾或學習問題,獨特的人格特徵,獨特的面部特徵以及心臟和血管(心血管)問題,可能與幾種基因的喪失有關。在這個地區。

雖然已經鑑定了一些與Williams綜合徵相關的特定基因,但是缺失區域中的大多數基因與Williams綜合徵的體徵和症狀之間的關係正在研究或未知。
染色體7的數量或結構的其他變化可導致生長和發育延遲,智力殘疾,獨特的面部特徵,骨骼異常,言語延遲和其他醫學問題。染色體7的變化包括每個細胞中來自該染色體的一些遺傳物質的額外拷貝(部分三體性7)或每個細胞中缺失的染色體片段(部分單體性7)。在一些情況下,幾個DNA構建塊(核苷酸)在染色體7的一部分中異常缺失或重複。稱為環染色體7的環狀結構也是可能的。
| 染色體異常型別 | ||||
▪1號染色體異常 ▪TAR綜合徵 ▪3p缺失綜合症 ▪嗜酸細胞性白血病 ▪貓叫綜合徵 ▪7q11.23重複綜合徵 ▪p11骨髓增殖綜合徵 ▪膀胱癌 ▪伊曼紐爾綜合症 ▪13號染色體異常 ▪多發性骨髓瘤 ▪小胖威利綜合徵 ▪20號染色體 | ▪1p36缺失綜合徵 ▪2號染色體異常 ▪3q29微缺失綜合徵 ▪4p綜合徵 ▪腦室周圍異位 ▪Silver綜合徵 ▪CBF-AML ▪Kleefstra綜合症 ▪尤因肉瘤 ▪視網膜細胞瘤 14號環形染色體 ▪16號染色體異常 ▪21號染色體 | ▪1q21.1微缺失 ▪2q37缺失綜合徵 ▪3q29微重複綜合徵 ▪5號染色體異常 ▪6號染色體異常 ▪SCS綜合徵 ▪8號染色體重組 ▪10號染色體異常 ▪雅克布森綜合症 ▪Patau綜合症 ▪15號染色體異常 ▪17號染色體 ▪22號染色體 | ▪1q21.1微重複綜合徵 ▪SATB2相關綜合徵 ▪4號染色體異常 ▪5q綜合徵 ▪6q24新生兒糖尿病 ▪威廉姆斯綜合徵 ▪毛髮鼻指綜合症 ▪11號染色體異常 ▪12號染色體異常 ▪14號染色體異常 ▪15q13.3微缺失 ▪18號染色體 | ▪神經母細胞瘤 ▪3號染色體異常 ▪肌營養不良症 ▪5q31.3微缺失綜合症 ▪7號染色體異常 ▪8號染色體異常 ▪9號染色體異常 ▪bws綜合症 ▪12p四體綜合徵 ▪FOXG1綜合症 ▪天使人綜合症 ▪19號染色體 |
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜