百科
文章
話題
脊柱骨骺發育不全
脊柱骨骺發育不全(SED)是一種罕見的遺傳病,根據亞型可以分為先天性脊柱骨骺發育不全、遲發性脊柱骨骺發育不全以及Wolcott-Rallison型脊柱骨骺發育不全。由於SED表現為長骨和椎體的骨骺發育異常並因此導致短軀幹侏儒,且此病有一定的遺傳率,因此建議有生育需求的患者通過第三代試管嬰兒技術助孕。
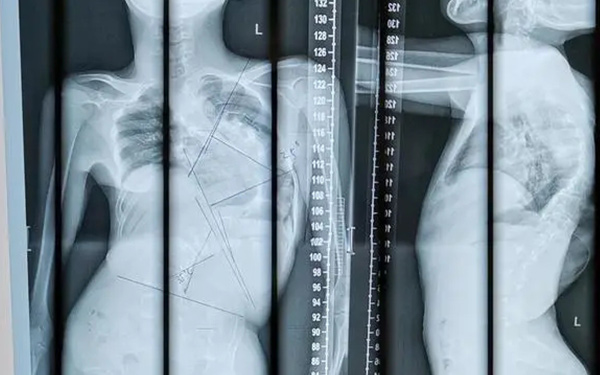
脊柱骨骺發育不全(spondyloepiphyseal dysplasia, SED)是一種罕見的遺傳病,發病率約為1/100 000,表現為長骨和椎體的骨骺發育異常並因此導致短軀幹侏儒,可以分為以下三種亞型:
先天性脊柱骨骺發育不全
臨床特徵:
出生時已可見胸骨異常,以後發展為胸廓明顯畸形。
兒童期呈短軀幹性侏儒,可伴有顎裂及畸形足。面部圓而扁平,頸短。腰椎過度前凸,脊椎側彎,齒狀突發育不良可造成寰樞關節不穩定。
晚期患者常出現寰椎錯位導致截癱。部分患者有近視和視網膜剝離,無角膜渾濁。智力正常。X線檢查可見:幹骺線不規則變寬,肋骨前端外展呈杯口狀。胸椎側位呈梨形(椎體後緣較前緣短),脊柱椎體扁平,椎間隙稍窄,齒狀突骨化不全。髂骨翼低,外展不足,髖臼頂呈水平狀,髖臼窩深。恥骨、恥骨頭及股骨頸骨化不全,長骨的骨骺扁平變形,以股骨頭最嚴重,手骨無明顯異常。
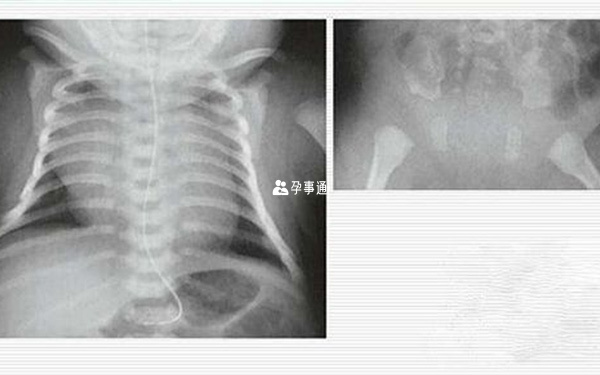
遲發性脊柱骨骺發育不全
臨床特徵:
出生和幼兒期無明顯症狀,多在5~10歲時出現生長緩慢,表現為身材矮小、短軀幹、桶狀胸、脊柱發育異常,形成短軀幹侏儒。髖及肩關節僅有輕度異常,腰背和四肢大關節(髖、肩)疼痛及活動受限。
X線檢查可見椎體變扁和椎間隙顯著狹窄,可有椎間盤鈣化。骨盆狹小、髂骨翼小。四肢大關節較早發生退行性病變。髖臼深,股骨頭扁,股骨頸短。
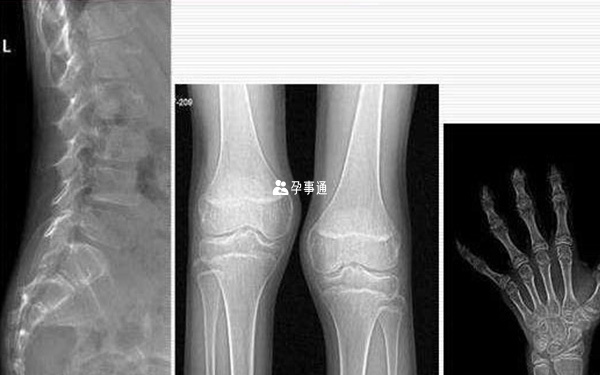
Wolcott-Rallison型脊柱骨骺發育不全
臨床特徵:
該病非常罕見,多發生在近親結婚較多的人群中。主要臨床表現有新生兒早發性糖尿病,骨骺發育異常,以及肝臟功能異常。骨骼異常主要影響長骨、骨盆和椎骨,而頭骨一般正常。膝部X光可見幹骺端變大且不規則,股骨和脛骨骨垢扁平,並伴有骨密度降低。
脊柱骨骺發育不全相關基因介紹
SEDC,常染色體顯性遺傳病,是由COL2A1基因(OMIM 120140)突變導致,定位與12q13.11,有57個外顯子。主要編碼合成Ⅱ型膠原蛋白,參與骨膜內成骨及軟骨內成骨的調控過程。其突變會造成Ⅱ型膠原蛋白結構異常,導致多種骨骼-軟骨發育異常疾病。突變型別包括單鹼基替換(錯義突變)、缺失和重複。突變後的蛋白產物會影響正常的螺旋結構並引起過度修飾,最終影響同源三聚體的形成。
SEDT, X連鎖隱性遺傳病,致病基因為TRAPPC2(OMIM 300202),該基因定位於Xp22.2。該基因編碼的蛋白產物SEDLIN含有140個氨基酸,是TRAPP(tracking prorein particle complex)的一個亞單位,參與蛋白質從內質網到高爾基體的運輸。目前發現的突變型別包括缺失突變、剪接位點突變、錯義突變和無義突變等。
Wolcott-Rallison型脊柱骨骺發育不全,常染色體隱性遺傳病,致病基因為EIF2AK3(PEK或PERK)(OMIM 604032),目前已發現EIF2AK3基因40種左右的突變型別,其中大部分為移碼突變或者無義突變,還包括錯義突變和剪接位點突變。在大多數家系中為純合突變,少數家系中為雙雜合突變。
案例分享
基本資訊
家系特點:父親為先天性脊柱骨骺發育不良(SEDC)患者,其COL2A1基因外顯子22位置存在c.1510G->A,p.G504S突變;母親正常;先證者為男方父親,為SEDC患者,男方母親正常。尋求PGD助孕。
檢測父母雙親、先證者2個,胚胎8個。
脊柱骨骺發育不全的檢測方法
三代試管嬰兒S-PGD——三重防護
晚髮型脊柱骨骺發育不全診斷方法
JBRH解決方案檢測特點
- 檢出率高,適用範圍廣;
- 檢測結果準確,能夠有效檢測到重組,可無先證者;
- 靶向捕獲設計,方案靈活,便於臨床成本控制。
脊柱骨骺發育不全診斷案例
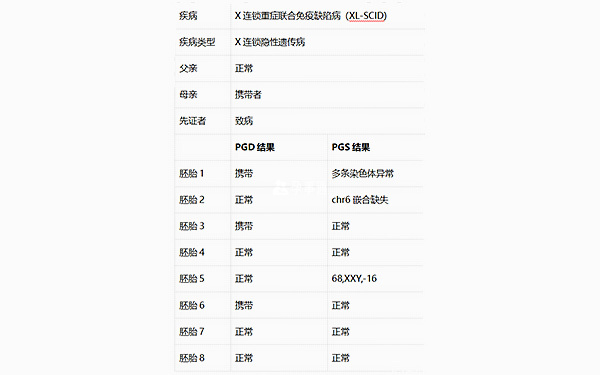
脊柱骨骺發育不全診斷結果
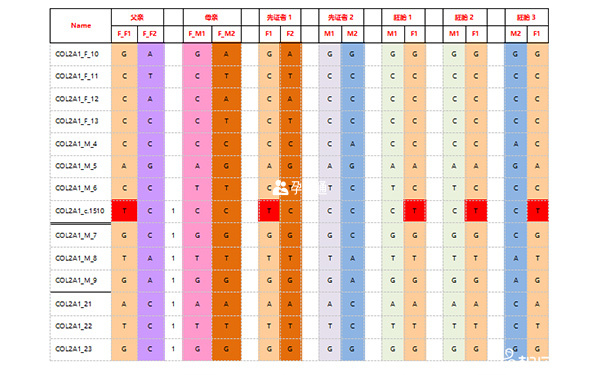
脊柱骨骺發育不全診斷結果2
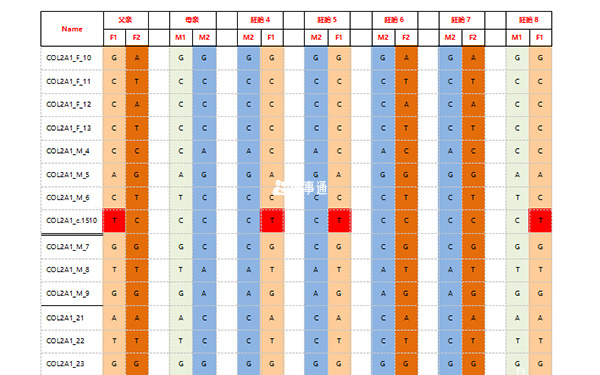
JBRH基於SNP連鎖分析的S-PGD解決方案優勢
脊柱骨骺發育不全做三代試管嬰兒的臨床意義:
- 更適用於臨床使用:對每種疾病精細化設計,檢測致病位點,可檢測到重組,檢測準確度更高;
- 分析結果更靈敏:測序不但能夠利用設計好的SNP,還能發現新的SNP並加以利用,提高了重組斷點的檢測靈敏度;
- 幫助減少先天性無虹膜症患兒的出生,有利於優生優育。
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜