百科
文章
話題
腎上腺腦白質
腎上腺腦白質營養不良是一種隱性遺傳性脂質代謝病,患此病的男性通常會有腦部病變、腎上腺髓神經病變、癲癇等症狀,女性攜帶者一般會有輕度到中度的痙攣性麻痺。由於該病在臨床上只能通過早發現早干預的方法予以緩解,無法有效治療,因此最佳的干預手段是通過第三代試管技術生育。
腎上腺腦白質營養不良是一種隱性遺傳性脂質代謝病,由於細胞中過氧化物酶體對超長鏈脂肪酸(VLCFA)的氧化發生障礙,以致VLCFA在血、腦白質、腎上腺皮質等器官和組織內大量聚積,引起中樞神經系統脫髓鞘和腎上腺皮質萎縮或發育不良。此外,還有注意力缺陷障礙(ADD)症狀的男孩還會表現出痴呆、進行性行為障礙、視力喪失、理解口語困難、書寫能力惡化、協調性障礙或其他神經障礙的跡象,包括進行性步態障礙、腿部僵硬或無力、括約肌控制異常、性功能障礙、伴有或不伴有腎上腺功能不全、認知或行為缺陷等。 而成年患病女性可能會患有進行性癱瘓,括約肌控制異常,感覺障礙主要影響雙腿。對於有ALD家族史的女性,診斷是基於臨床特徵(最常見的進展性痙攣性麻痺)和一系列實驗室檢測(包括ABCDI基因檢測)。

導致腎上腺腦白質營養不良的原因
致病基因ABCDl(ATP-binding Cassette,Sub-family D,Memberl)位於X染色體q28區域,由10個外顯子和9個內含子組成,編碼1個含745個氨基酸殘基的蛋白質,定義為腎上腺腦白質營養不良蛋白(adrenoleukodystrophy protein,ALDP),位於過氧化物酶體膜上。ALDP和另外3個位於過氧化物酶體膜上的蛋白結合,形成二聚體,能將飽和的VLCFA轉運至過氧化物酶體內進行氧化。由於ABCDl基因突變,導致ALDP功能異常,使得VLCFA的氧化受阻,引起VLCFA聚集。VLCFA在神經系統中聚集可破壞髓鞘的正常形成和髓鞘的穩定性,在腎上腺皮質細胞中聚集可引起腎上腺皮質細胞膜表面的ACTH受體功能下降,細胞內類固醇合成受抑制,而致腎上腺功能減退。
流行病學
患病率為新生男嬰1/2萬,女嬰為1/5萬
腎上腺腦白質營養不良的症狀
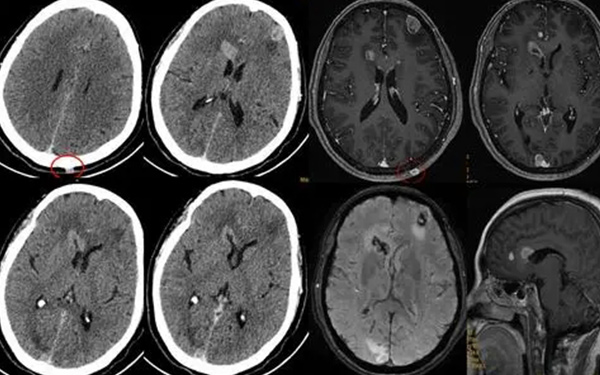
ALD的病理特點是通過影像學檢查可見腦白質病變區呈固狀或橡膠狀、腎上腺皮質萎縮但髓質不受影響,此外可以發現腦組織、腎上腺、周圍神經、睪丸內有特徵性的包容體,包容體內富含VLCFA 酯化的膽固醇。腦部最顯著的特徵是沿大腦前後軸由枕向額頂方向發展的脫髓鞘性改變,故在任何情況下,總是枕區腦白質受累最重,而U型纖維保留;另一特徵是血管周圍的炎性細胞浸潤。
此外,x-腎上腺腦白質營養不良症(X-ALD)的表型表達範圍很廣,因此不能通過VLCFA水平或家族史來簡單評估,有些X-ALD患者甚至到成年以後依舊不會發病。
最常見的男性症狀:
症狀1:腦部病變(>70%)
發病最常發生在4-8歲之間,在7歲時達到高峰。致病的男孩表現出行為或學習上的缺陷,通常被診斷為注意力缺陷障礙或多動症,但可以通過藥物治療予以控制。隨後出現更嚴重的潛在症狀,包括注意力不集中、書寫技能下降、語言理解困難、閱讀困難、空間定位困難、閱讀理解困難、視覺障礙,偶爾複視;以及攻擊性或去抑制的行為。此時進行的腦MRI檢查也可能異常顯著。 儘管情況因人而異,但進展速度很快,在0.5-2年內完全喪失行為能力,大約40%-45%的患者表現出一定程度的腦損傷。20%的患者大腦受累變得非常嚴重,並導致嚴重的認知和行為障礙,進而可能導致完全殘疾和死亡。
症狀2:腎上腺髓神經病變(40-45%)
大多數人在第一次注意到神經紊亂時,腎上腺皮質功能受損。在二十多歲時,出現雙腿逐漸僵硬無力、括約肌控制異常、性功能障礙等,所有的症狀都是逐漸加重的。大約70%患者在首次出現神經症狀時腎上腺皮質功能受損,嚴重者會演變為腎上腺髓神經病變(AMN)。
症狀3:癲癇(約為10%)
在一些男孩患者中,癲癇可能是最先出現的症狀。其餘可能伴隨出現的症狀包括頭痛、顱內壓增高、偏癱或視野缺損、失語或其他侷限性腦疾病的徵象,發病年齡通常在4歲到10歲之間。

女性攜帶者出現的症狀:
超過20%的女性攜帶者在中年或以後出現輕度到中度的痙攣性麻痺,但腎上腺功能正常。
檢測和診斷
臨床上通過以下:
- 1. 神經檢查;
- 2. 大腦核磁共振;
- 3. 腎上腺功能測試;
- 4. 基因檢測;
- 5. 遺傳諮詢。
X-ALD的診斷是建立在男性患者臨床發現超高的超長鏈脂肪酸(VLCFA)水平,測量VLCFA足以確定大多數患病男性的X-ALD診斷。當VLCFA檢測結果不確定時,需要通過分子遺傳學檢測半合子ABCD1基因突變來確診。而女性攜帶者則需要通過基因檢測檢查ABCD1基因突變和VLCFA是否升高來判斷。
治療方法
目前臨床上只能通過早發現早干預的方法予以緩解,無法有效治療。治療主要包括皮質類固醇替代治療(聯合洛伐他汀)、評估腎上腺皮質功能、檢測腦部狀況等,但對於腦部出現的病變則無能為力(有醫生嘗試用環磷醯胺和免疫球蛋白治療,但未見療效)。
其它治療方面,目前骨髓移植和基因治療是熱門話題,但依舊無法徹底解決腦部病變,一旦發生腦部病變,預後極差,一般出現神經症狀後1-3年死亡。

遺傳規律
X-ALD以x-連鎖隱性遺傳的方式遺傳,約95%的患者從父母一方遺傳了ABCD1致病基因,其餘5%患者是自發性的基因突變。一般男性患者的後代如果是女兒則會成為疾病攜帶者,是兒子則不會致病;女性攜帶者的後代如果是兒子,有50%致病,如果後代是女兒則有50%的機率成為攜帶者。因此,及早通過基因檢測確診家族遺傳病史至關重要,這會直接影響到後續的遺傳諮詢和泰國試管嬰兒程序。
能不能避免遺傳
遺傳諮詢是向ALD家庭提供有關遺傳疾病的性質、遺傳和影響的資訊,以幫助他們作出明智的醫療選擇和生育選擇的過程,如果夫妻雙方有患有ALD疾病,那麼及早確定遺傳風險、澄清攜帶者狀態和討論產前檢測的最佳時間是在懷孕前,而最佳的干預手段則是泰國試管嬰兒技術(IVF),尤其是三代試管嬰兒技術(PGD)。
胚胎植入前基因檢查(PGD),主要用於檢查胚胎是否攜帶有遺傳缺陷的基因。它是在試管嬰兒技術基礎上出現的,精子卵子在體外結合形成受精卵,併發育成胚胎後,要在其植入子宮前進行基因檢測,以便使體外授精的試管嬰兒避免一些遺傳疾病。一旦在基因檢測中發現了家族成員中有ABCD1基因突變,就需要對打算懷孕的女性進行PGD檢查,從根源上徹底阻斷該病遺傳給後代(尤其是胚胎檢測發現是男嬰時)。
而ALD屬於X連鎖隱性遺傳疾病,泰國試管嬰兒技術可以通過對胚胎特定的致病基因ABCD1進行檢測以篩查特定的疾病(比如X-ALD)。而且通過篩選出的優質胚胎在子宮著床的機率更大,更低的自然死亡率,同時也減低了生出有異常染色體的後代的風險,是ALD患者家庭最理想的干預手段。
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜