百科
文章
話題
麥克綜合徵
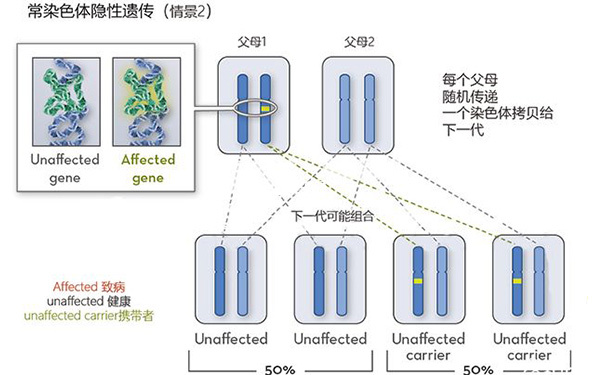
Meckel綜合徵是一種致命且罕見的常染色體隱性遺傳病,其特徵是枕部腦膨出三聯徵、大多囊腎和後交叉多發性。目前診斷Meckel-Gruber綜合徵的最佳方法產前超聲檢查,不過隨著超聲與分子診斷等技術的發展,Meckel-Gruber綜合徵患者可以選擇通過胚胎植入前診斷技術篩查出健康的胚胎並移植。
導航
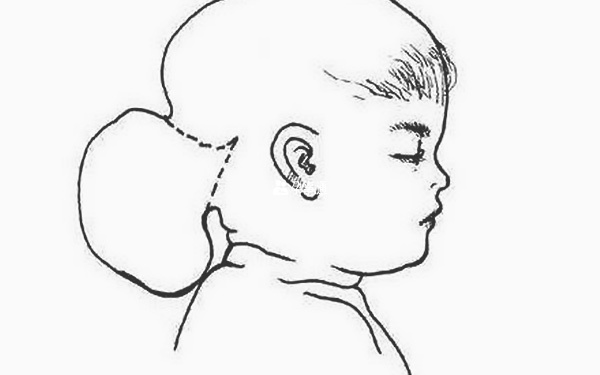
Meckel-Gruber綜合徵(MKS)是一種致命的,罕見的常染色體隱性遺傳病,其特徵是枕部腦膨出三聯徵,大多囊腎和後交叉多發性。別名:腦膨出、多指、多囊腎綜合徵、Meckel綜合徵、Glubef綜合徵。相關異常包括口腔切割 ; 生殖器異常; 中樞神經系統(CNS)畸形,包括Dandy-Walker和Arnold-Chiari畸形; 和肝纖維化。肺發育不全是導致死亡的主要原因。早在妊娠10周時,超聲檢查的改進就可以進行產前診斷。
遺傳模式
關於Meckel-Gruber綜合症的第一份報告由Johann Friedrich Meckel於1822年發表。GB Gruber還在1934年發表了Meckel-Gruber綜合徵患者的報告,並將其命名為dysencephalia splanchnocystica。Meckel-Gruber綜合徵也稱為Meckel綜合徵或Gruber綜合徵。
條件(OMIM 24900)已被對映到染色體17q21-24(MKS1)12不同基因座,11q13(MKS2),8q21.3-q22.1(MKS3), 12q21.31-q21.33 (MKS4),16q12.2(MKS5),4p15.3(MKS6), 3q22.1(MKS7), 12q24.31(MKS8), 17p11.2 (MKS9), 19q13.2(MKS10),16q23.1(MKS11),和1q32.1(MKS12)。 這種對映表明Meckel-Gruber綜合徵的遺傳異質性。
Barisic等人使用來自191例Meckel-Gruber綜合徵的資料進行的一項研究,通過歐洲先天性異常監測(EUROCAT)網路獲得,發現該疾病的各種特徵的流行程度如下 :
囊性腎(97.7%) 多指(87.3%) 腦膨出(83.8%) 肝臟的纖維化/囊性變化(65.5%,通過屍檢檢查確定) 其他CNS異常(51.4%) 口面裂隙(31.8%) 後處理
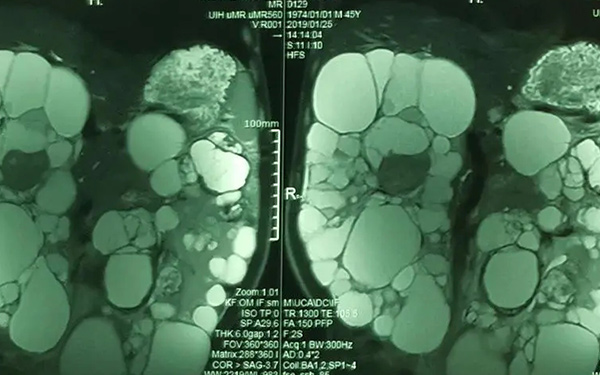
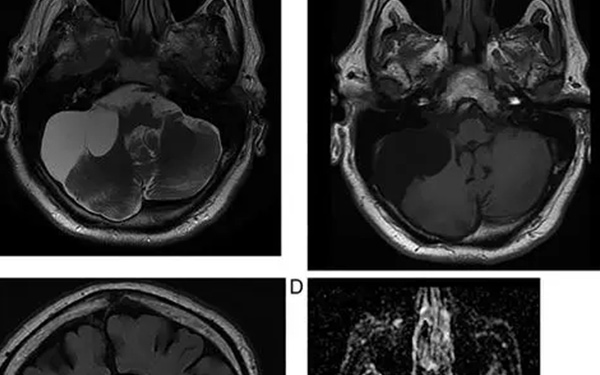
產前超聲檢查是目前診斷Meckel-Gruber綜合徵的最佳方法,可用於二維(2D),三維(3D)和四維(4D)模式。
在存在嚴重羊水過少的情況下,磁共振成像(MRI)是評估胎兒異常的超聲檢查的有價值的補充。它主要用於超聲檢查結果不確定或不足以指導治療選擇時。
染色體分析對於排除13號三體性是必不可少的,Meckel-Gruber綜合徵模仿該三體性。
如果在妊娠早期早期檢測到異常,如果羊水過少不允許羊膜腔穿刺,則可以在妊娠10-12周或妊娠後期進行絨毛膜絨毛取樣(CVS)。如果存在足夠的液袋,則在妊娠14周後進行羊膜穿刺術。
Meckel-Gruber綜合徵患者可能需要對腦膨出進行心臟修復或神經外科干預。
通過三代試管嬰兒正常生育
隨著超聲與分子診斷等技術的發展,尤其是第三代試管嬰兒技術(胚胎植入前診斷技術)的出現使解決疾病基因攜帶者的生育問題出現了曙光,Meckel-Gruber綜合徵患者可以通過胚胎植入前診斷技術篩查出健康的胚胎進行移植。
四代試管
卵巢早衰交流群三代試管
優生優育攻略群高齡生子
絕經助孕攻略群無精/死精
顯微取精交流群冷凍卵子
生育力儲存經驗群出國生子
海外試管交流群大家都在搜